Why Johnny can’t read (but Jane can)
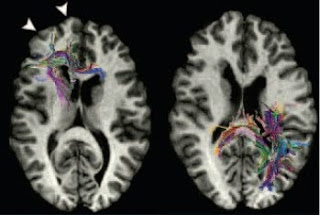
Reading is not a skill that comes naturally. Unlike learning spoken language, which the human brain has evolved to absorb almost effortlessly, learning to read is a protracted and difficult process. It involves the categorical association of arbitrary visual symbols with phonemes and also the ability to break words down into component phonemes. It thus relies on an integration between visual and auditory processes, combining spatial and temporal information, within a learned linguistic context. The fact that reading is such a specialized and integrative skill may partly explain why it can be selectively impaired in people of otherwise normal intellectual abilities. Dyslexia, as this type of selective reading difficulty is called, is quite common, affecting anywhere from 5% to 20% of children, depending on the criteria used in its diagnosis. Cohort studies which directly tested all individuals have found that dyslexia is about twice as common in boys as in girls. This is n...