Split brains, autism and schizophrenia
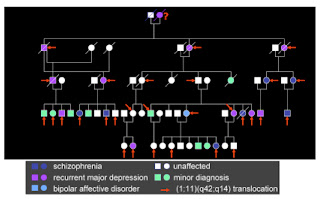
A new study suggests that a gene known to be causally linked to schizophrenia and other psychiatric disorders is involved in the formation of connections between the two hemispheres of the brain. DISC1 is probably the most famous gene in psychiatric genetics, and rightly so. It was discovered in a large Scottish pedigree, where 18 members were affected by psychiatric disease.
The diagnoses ranged from schizophrenia and bipolar disorder to depression and a range of “minor” psychiatric conditions. It was found that the affected individuals had all inherited a genetic anomaly – a translocation of genetic material between two chromosomes. This basically involves sections of two chromosomes swapping with each other. In the process, each chromosome is broken, before being spliced back to part of the other chromosome. In this case, the breakpoint on chromosome 1 interrupted a gene, subsequently named Disrupted-in-Schizophrenia-1, or DISC1.
That this discovery was made using cl...