Visualising Connections in the Human Brain
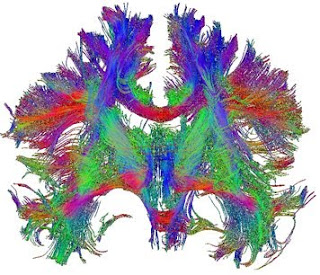
Most of us are familiar with pictures from magnetic resonance imaging (MRI) of the human brain; indeed, these black-and-white images have achieved almost iconic status at this stage. From popular television programmes, and regrettably from common experience, the use of these images to detect lesions, such as tumours or the effects of stroke, is well known. Classic MRI can distinguish grey and white matter based on their different cellular composition but cannot go very far beyond that, because all the white matter has effectively the same contrast. This makes traditional MRI of limited use in examining connectivity between areas of the brain, except at a very gross level (such as whether the corpus callosum exists, for example). However, with a few modifications, MRI can be applied to non-invasively interrogate connectivity in the living human brain, with ever-increasing sensitivity. These new techniques are opening up avenues of investigation that have not just tremendous clinical...