Self-organising principles in the nervous system

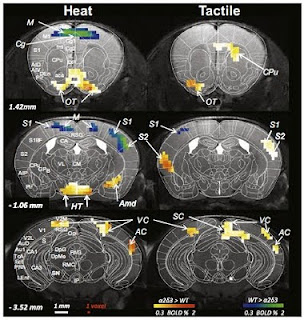
The circuitry of the brain is too complex to be completely specified by genetic information – at least not down to the level of each connection. There are hundreds of billions of neurons in your brain, each making an average of 1,000 connections to other cells. There are simply not enough genes in the genome to specify all of these connections. What the genetic program can achieve is a very good wiring diagram of initial projections between neurons in different brain areas (or layers or between particular cell types). This circuitry is then refined and elaborated at the cellular level by processes of activity-dependent development, under the principle that “cells that fire together, wire together”. The circuitry of the brain is thus a self-organising system, which assembles under the influence of local interactions, mediated first by molecular interactions and second by patterns of electrical activity. A new study highlights an important additional factor that allows global patt...