A synaesthetic mouse?
An amazing study just published in Cell starts out with fruit flies insensitive to pain and ends up with what looks very like a synaesthetic mouse. Penninger and colleagues were interested in the mechanisms of pain sensation and have been using the fruit fly as a model to investigate the underlying biological processes. Like any good geneticist faced with profound ignorance of how a process works, they began by screening for mutant flies that are insensitive to pain. Making use of a very powerful genetic resource developed in Vienna (a bank of fly lines expressing RNA interference constructs for every gene in the genome) they screened through all these genes to see which ones were required in neurons for flies to respond to pain. (In particular, pain caused by excessive heat).
Why should anyone care how a fly feels pain? Well, like practically everything else you can think of, the basic physiology and molecular biology of pain sensation is very highly conserved from flies to mammals. It starts with specialized proteins called TRP channels, which are ion channels that span the cell membrane and allow ions to pass across it in response to various stimuli. Some of these TRP channels respond specifically to painful stimuli, some even more specifically to painful heat, and these molecules are highly conserved. The hope was that by screening for other genes they would identify additional conserved elements of the pathway.
This was exactly what they found. Among hundreds of new mutants that were insensitive to pain, they focused in this report on one, a gene called straightjacket. This gene codes for a protein called alpha2delta3, or CACNA2D3, which is a member of a conserved family of proteins that make up part of a calcium channel. These proteins are involved in modulating neurotransmission and also in some aspects of development, including the formation of synapses. Interestingly, mutations in other members of this gene family are associated with bipolar disorder, schizophrenia, Timothy syndrome (the symptoms of which include autism), epilepsy and migraine.
This particular gene is conserved in mammals and the authors show that mutation of the gene in mice also leads to insensitivity to pain induced by heat, but not to painful mechanical stimuli – a remarkably specific functional conservation. In addition, they show suggestive evidence that variants in the gene in humans are also associated with a higher pain tolerance. These latter data will have to be replicated but tantalizingly suggest that variation in this gene in humans may contribute to differences in pain sensitivity.
Mutation of this gene seems to cause pain insensitivity not by blocking pain responses in the sensory neurons or by blocking transmission of this signal to the brain, but by blocking transmission from the first relay station of the brain, the thalamus, to the cortex, where it must pass to be consciously perceived. The authors could show that the sensory neurons still respond to painful stimuli and that a spinal pain reflex was intact. They also used functional magnetic resonance imaging in mice to show that the thalamus was active as normal in response to painful stimuli. However, a network of areas in the cortex (the “pain matrix”) was completely unresponsive. Somehow, deletion of CACNA2D3 alters connectivity within the thalamus or from thalamus to cortex in a way that precludes transmission of the signal to the pain matrix areas.
This is where the story really gets interesting. While they did not observe responses of the pain matrix areas in response to painful stimuli, they did observe something very unexpected – responses of the visual and auditory areas of the cortex! What’s more, they observed similar responses to tactile stimuli administered to the whiskers. Whatever is going on clearly affects more than just the pain circuitry.
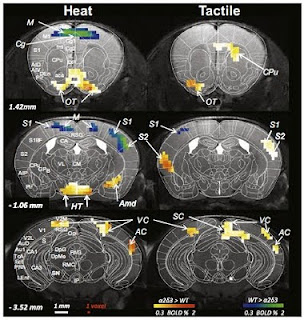
The authors suggest that this kind of sensory cross-activation may represent a model for synaesthesia, which is characterised by very similar effects. While this condition is highly familial, no genes have yet been isolated for it. Could CACNA2D3 be a viable candidate? It certainly seems possible, though one point suggests that whatever is happening, while similar to developmental synasthesia, may be somewhat distinct.
Synaesthesia usually involves an extra percept in response to some stimulus, without any decrement in the response to the stimulus itself. So, people who see colours when they hear music hear the music normally – the colour is just part of that experience. This is rather different from a situation where one sense is deficient and is taken over by another. That situation can arise due to injury, for example, and can even be surgically induced in animal models (used to study brain plasticity). One recent report (see below) described a patient who had a lesion in the thalamus in the somatosensory nucleus. This region was subsequently invaded by fibres carrying auditory information so that the patient was able to feel sounds. (The auditory fibres were activated by sound, which cross-activated the somatosensory area, which communicated this activity to the somatosensory cortex, where it was perceived as a touch on the surface of the body).
Could such an effect explain what was happening in these mice? Perhaps for the pain circuits, though one would typically expect that they would be invaded by other senses, rather than the other way around. But for the tactile stimuli, the message was apparently still getting through to the somatosensory cortex, it was just also activating visual and auditory areas. That starts to look like a pretty good model for synaesthesia. Whether it really is would most convincingly be demonstrated by finding a mutation in this gene in someone with synaesthesia. A good place to start might be testing the carriers of the variants in this gene in humans which affected pain sensitivity for any signs of synaesthesia.
Even if it does not correspond exactly to what we call developmental synaesthesia, one can predict that something pretty strange would result from mutation of this gene in humans. Given that every base of the genome is probably mutant in someone on the planet it seems certain that such mutations will eventually crop up.
It is not yet clear what cellular mechanism can explain the cross-activation observed in the mutant mice. One can imagine any number of scenarios, including structural rewiring between thalamic nuclei (which are specialized to transmit different types of sensory information) or from thalamus to cortex. Alternatively, changes in neurotransmission might explain the effects, for example by damping down cross-inhibitory processes that normally sharpen responses to one sense at a time. One way to dissociate these would be to see whether blocking the function of the protein just in adults is sufficient to induce the effect or if it has to be blocked during development. This might be achieved using drugs – a close relative of CACNA2D3 is blocked by gabapentin, a drug used in humans as an antiepileptic and also to block neuropathic pain (like that which can arise due to shingles, for example). Whether this or a similar drug could affect the A2D3 subunit is not, I think, known, but no doubt someone is now looking for a drug that can.
Neely GG, Hess A, Costigan M, Keene AC, Goulas S, Langeslag M, Griffin RS, Belfer I, Dai F, Smith SB, Diatchenko L, Gupta V, Xia CP, Amann S, Kreitz S, Heindl-Erdmann C, Wolz S, Ly CV, Arora S, Sarangi R, Dan D, Novatchkova M, Rosenzweig M, Gibson DG, Truong D, Schramek D, Zoranovic T, Cronin SJ, Angjeli B, Brune K, Dietzl G, Maixner W, Meixner A, Thomas W, Pospisilik JA, Alenius M, Kress M, Subramaniam S, Garrity PA, Bellen HJ, Woolf CJ, & Penninger JM (2010). A Genome-wide Drosophila Screen for Heat Nociception Identifies α2δ3 as an Evolutionarily Conserved Pain Gene. Cell, 143 (4), 628-38 PMID: 21074052
Beauchamp MS, & Ro T (2008). Neural substrates of sound-touch synesthesia after a thalamic lesion. The Journal of neuroscience : the official journal of the Society for Neuroscience, 28 (50), 13696-702 PMID: 19074042
Why should anyone care how a fly feels pain? Well, like practically everything else you can think of, the basic physiology and molecular biology of pain sensation is very highly conserved from flies to mammals. It starts with specialized proteins called TRP channels, which are ion channels that span the cell membrane and allow ions to pass across it in response to various stimuli. Some of these TRP channels respond specifically to painful stimuli, some even more specifically to painful heat, and these molecules are highly conserved. The hope was that by screening for other genes they would identify additional conserved elements of the pathway.
This was exactly what they found. Among hundreds of new mutants that were insensitive to pain, they focused in this report on one, a gene called straightjacket. This gene codes for a protein called alpha2delta3, or CACNA2D3, which is a member of a conserved family of proteins that make up part of a calcium channel. These proteins are involved in modulating neurotransmission and also in some aspects of development, including the formation of synapses. Interestingly, mutations in other members of this gene family are associated with bipolar disorder, schizophrenia, Timothy syndrome (the symptoms of which include autism), epilepsy and migraine.
This particular gene is conserved in mammals and the authors show that mutation of the gene in mice also leads to insensitivity to pain induced by heat, but not to painful mechanical stimuli – a remarkably specific functional conservation. In addition, they show suggestive evidence that variants in the gene in humans are also associated with a higher pain tolerance. These latter data will have to be replicated but tantalizingly suggest that variation in this gene in humans may contribute to differences in pain sensitivity.
Mutation of this gene seems to cause pain insensitivity not by blocking pain responses in the sensory neurons or by blocking transmission of this signal to the brain, but by blocking transmission from the first relay station of the brain, the thalamus, to the cortex, where it must pass to be consciously perceived. The authors could show that the sensory neurons still respond to painful stimuli and that a spinal pain reflex was intact. They also used functional magnetic resonance imaging in mice to show that the thalamus was active as normal in response to painful stimuli. However, a network of areas in the cortex (the “pain matrix”) was completely unresponsive. Somehow, deletion of CACNA2D3 alters connectivity within the thalamus or from thalamus to cortex in a way that precludes transmission of the signal to the pain matrix areas.
This is where the story really gets interesting. While they did not observe responses of the pain matrix areas in response to painful stimuli, they did observe something very unexpected – responses of the visual and auditory areas of the cortex! What’s more, they observed similar responses to tactile stimuli administered to the whiskers. Whatever is going on clearly affects more than just the pain circuitry.

The authors suggest that this kind of sensory cross-activation may represent a model for synaesthesia, which is characterised by very similar effects. While this condition is highly familial, no genes have yet been isolated for it. Could CACNA2D3 be a viable candidate? It certainly seems possible, though one point suggests that whatever is happening, while similar to developmental synasthesia, may be somewhat distinct.
Synaesthesia usually involves an extra percept in response to some stimulus, without any decrement in the response to the stimulus itself. So, people who see colours when they hear music hear the music normally – the colour is just part of that experience. This is rather different from a situation where one sense is deficient and is taken over by another. That situation can arise due to injury, for example, and can even be surgically induced in animal models (used to study brain plasticity). One recent report (see below) described a patient who had a lesion in the thalamus in the somatosensory nucleus. This region was subsequently invaded by fibres carrying auditory information so that the patient was able to feel sounds. (The auditory fibres were activated by sound, which cross-activated the somatosensory area, which communicated this activity to the somatosensory cortex, where it was perceived as a touch on the surface of the body).
Could such an effect explain what was happening in these mice? Perhaps for the pain circuits, though one would typically expect that they would be invaded by other senses, rather than the other way around. But for the tactile stimuli, the message was apparently still getting through to the somatosensory cortex, it was just also activating visual and auditory areas. That starts to look like a pretty good model for synaesthesia. Whether it really is would most convincingly be demonstrated by finding a mutation in this gene in someone with synaesthesia. A good place to start might be testing the carriers of the variants in this gene in humans which affected pain sensitivity for any signs of synaesthesia.
Even if it does not correspond exactly to what we call developmental synaesthesia, one can predict that something pretty strange would result from mutation of this gene in humans. Given that every base of the genome is probably mutant in someone on the planet it seems certain that such mutations will eventually crop up.
It is not yet clear what cellular mechanism can explain the cross-activation observed in the mutant mice. One can imagine any number of scenarios, including structural rewiring between thalamic nuclei (which are specialized to transmit different types of sensory information) or from thalamus to cortex. Alternatively, changes in neurotransmission might explain the effects, for example by damping down cross-inhibitory processes that normally sharpen responses to one sense at a time. One way to dissociate these would be to see whether blocking the function of the protein just in adults is sufficient to induce the effect or if it has to be blocked during development. This might be achieved using drugs – a close relative of CACNA2D3 is blocked by gabapentin, a drug used in humans as an antiepileptic and also to block neuropathic pain (like that which can arise due to shingles, for example). Whether this or a similar drug could affect the A2D3 subunit is not, I think, known, but no doubt someone is now looking for a drug that can.
Neely GG, Hess A, Costigan M, Keene AC, Goulas S, Langeslag M, Griffin RS, Belfer I, Dai F, Smith SB, Diatchenko L, Gupta V, Xia CP, Amann S, Kreitz S, Heindl-Erdmann C, Wolz S, Ly CV, Arora S, Sarangi R, Dan D, Novatchkova M, Rosenzweig M, Gibson DG, Truong D, Schramek D, Zoranovic T, Cronin SJ, Angjeli B, Brune K, Dietzl G, Maixner W, Meixner A, Thomas W, Pospisilik JA, Alenius M, Kress M, Subramaniam S, Garrity PA, Bellen HJ, Woolf CJ, & Penninger JM (2010). A Genome-wide Drosophila Screen for Heat Nociception Identifies α2δ3 as an Evolutionarily Conserved Pain Gene. Cell, 143 (4), 628-38 PMID: 21074052
Beauchamp MS, & Ro T (2008). Neural substrates of sound-touch synesthesia after a thalamic lesion. The Journal of neuroscience : the official journal of the Society for Neuroscience, 28 (50), 13696-702 PMID: 19074042



Very interesting. It's too bad we can't ask the mice whether they saw or heard things when given noxious stimulation. It would be interesting if upcoming work on the genetics of synesthesia identifies involvement of the straightjacket gene. Although I'm not sure how common it is in humans that painful stimulation elicits vision or sound.
ReplyDeleteColoured pain is a fairly common form of synaesthesia actually (about 5% of synaesthetes, according to Sean Day's excellently curated website):
ReplyDeletehttp://home.comcast.net/~sean.day/html/types.html
I have heard reports of pain inducing sounds but that is much more rare.
A very exciting thing I can tell you. I would have liked a little more so sites. Keep it up guys.
ReplyDeletecustom essay paper
This is so amazing to see how people think this way so much here. So many go different ways on this so much here. So good to learn about here. Great job on this. Cliquez-ici
ReplyDeleteNice post with awesome points! Can’t wait for the next one.
ReplyDeleteMSI - 11.6" Touch-Screen Laptop - 8GB Memory - 128GB Solid State Drive
MSI - 15.6" Laptop - 12GB Memory - 750GB Hard Drive