Nerves of a feather, wire together
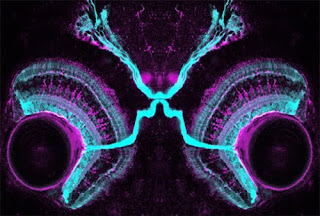
Finding your soulmate, for a neuron, is a daunting task. With so many opportunities for casual hook-ups, how do you know when you find “the one”? In the early 1960’s Roger Sperry proposed his famous “chemoaffinity theory” to explain how neural connectivity arises. This was based on observations of remarkable specificity in the projections of nerves regenerating from the eye of frogs to their targets in the brain. His first version of this theory proposed that each neuron found its target by expression of matching labels on their respective surfaces. He quickly realised, however, that with ~200,000 neurons in the retina, the genome was not large enough to encode separate connectivity molecules for each one. This led him to the insight that a regular array of connections of one field of neurons (like the retina) across a target field (the optic tectum in this case) could be readily achieved by gradients of only one or a few molecules. The molecules in question, Ephrins and Eph rec...
