From miswired brain to psychopathology – modelling neurodevelopmental disorders in mice
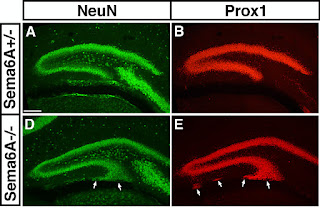
It takes a lot of genes to wire the human brain. Billions of cells, of a myriad different types have to be specified, directed to migrate to the right position, organised in clusters or layers, and finally connected to their appropriate targets. When the genes that specify these neurodevelopmental processes are mutated, the result can be severe impairment in function, which can manifest as neurological or psychiatric disease. How those kinds of neurodevelopmental defects actually lead to the emergence of particular pathological states – like psychosis or seizures or social withdrawal – is a mystery, however. Many researchers are trying to tackle this problem using mouse models – animals carrying mutations known to cause autism or schizophrenia in humans, for example. A recent study from my own lab ( open access in PLoS One ) adds to this effort by examining the consequences of mutation of an important neurodevelopmental gene and providing evidence that the mice...